 To get started you don't have to get involved with the scattering rules. Your job (or actually your computer's job) is to interpret the spectral information and refer it against established spectral library of known materials. Without a reference library the Raman spectra are just a set of wiggly lines.
To get started you don't have to get involved with the scattering rules. Your job (or actually your computer's job) is to interpret the spectral information and refer it against established spectral library of known materials. Without a reference library the Raman spectra are just a set of wiggly lines.
Raman shift is too weak effect to be observed by bare eyes. High sensitivity digital spectrometer is needed to resolve the "emission lines" instead of a traditional spectroscope.
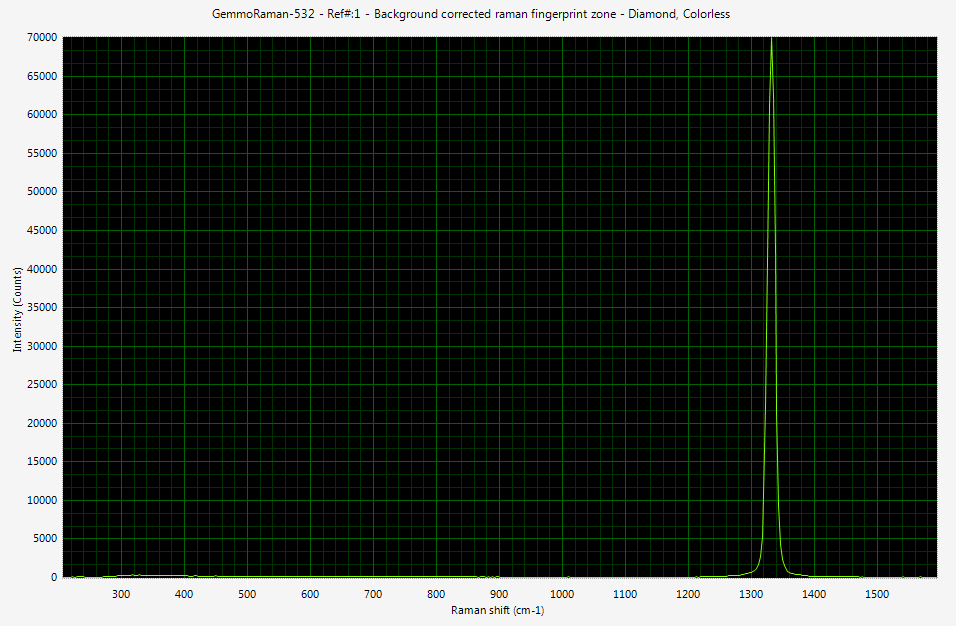
On the right side you can see Raman spectrum of colorless diamond recorded with digital spectrometer. Please click the image to see it in full size. The x-axis of spectrum contains the wavelength information (don't let cm-1 units to bother your, these are discussed later) and y axis is the intensity of light.
The diamond Raman peak produced by sp3 bonding between carbon atoms is located at 1332 cm-1. The excitation source in this case was monochromatic 532 nm (green) laser. The source has to be monochromatic and powerful enough to create detectable amount of shifted photons. In real life laser is the only light source fulfilling these requirements.